µĄģĶ░ł’╝Üń½ŗÕ╝éĶŹ»ńē®INDńö│Ķ»Ęõ╣ŗĶŹ»ÕŁ”ńĀöń®Č
ń½ŗÕ╝éĶŹ»ńē®ńÜäÕ╝ĆÕÅæµś»õĖĆõĖ¬µ×üÕģʵÄóń┤óµĆ¦ńÜäńĀöń®ČÕÄåń©ŗ�’╝ī�’╝īÕģČķĆÜÕĖĖńö▒µ£¬ń¤źµ£ĆÕģł�’╝ī�’╝īÕ¤║õ║ĵ£¬Ķó½ń¤źĶČ│ńÜäõĖ┤Õ║Ŗķ£Ćµ▒é�’╝ī�’╝īÕÄ╗Õ╝ĆÕ▒ĢĶŹ»ńē®ńŁøķĆēõĖÄÕÅæµśÄńÜäõ║ŗµāģ�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕĘ«Õł½õ║Äõ╗┐ÕłČĶŹ»�’╝ī�’╝īń½ŗÕ╝éĶŹ»ńē®ńÜäńĀöń®Č�’╝ī�’╝īµś»ķÜÅńØĆÕĘ«Õł½ķśČµ«ĄĶĆīķĆɵŁźµĘ▒ÕģźńØüÕ╝ĆńÜä�’╝ī�’╝īµ»ÅķśČµ«ĄńĀöń®ČńÜäµĘ▒Õ║”ķĆÜ�’╝ø’╝ø�’╝øÕ▓½µØ鎳ńĢöńģŖ╬”ń║│ń¼āÕ¦źÕ│ĪńĪŁ�’╝ī�’╝īõ╗ÄĶĆīķś╗µŁóõĖŹķĪ╗Ķ”üńÜäÕż¬Ķ┐ćÕ╝ĆÕÅæ�’╝ī�’╝īõ╗źĶŖéń║”ÕÉäµ¢╣ķØóńÜäĶĄäµ║É�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕ£©µłæÕøĮ�’╝ī�’╝īń½ŗÕ╝éĶŹ»ńē®ńÜäÕ╝ĆÕÅæ�’╝ī�’╝īĶÖĮĶĄĘµŁźĶŠāµÖÜõĖöÕ░ÜõĖŹÕÅ»ńå¤�’╝ī�’╝īõĮåµĢ┤õĮōĶČŗÕŖ┐µŁŻÕ£©Ķ┐ĮĶĄČĶź┐µ¼¦µŚź�’╝ī�’╝īµ»ÅõĖ¬ńÄ»ĶŖéń╗åĶŖéõ╣¤ķāĮÕ£©Õ«īÕ¢äÕĮōõĖŁ�’╝ī�’╝īÕ”éõĖŗķØóĶ”üĶüŖńÜäINDńö│Ķ»ĘõĖŁńÜäŌĆ£ĶŹ»ÕŁ”ńĀöń®ČŌĆØ�’╝ī�’╝īÕ░▒µś»ÕģČõĖŁõ╣ŗõĖĆ�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
ńŠÄÕøĮFDA&ń½ŗÕ╝éĶŹ»INDķśČµ«Ą-ĶŹ»ÕŁ”ńĀöń®Č
1. FDAÕģ│õ║ÄINDķśČµ«ĄĶŹ»ńē®ńÜäÕ«ĪĶ»äµīćÕŹŚ
µŚ®Õ£©1995Õ╣┤�’╝ī�’╝īńŠÄÕøĮķŻ¤ńē®ĶŹ»ÕōüńøæĶ¦åµ▓╗ńÉåÕ▒ĆńÜäCDERÕÆīCBERõŠ┐Õ«ŻÕĖāõ║åÕģ│õ║ÄINDķśČµ«ĄńÜäĶŹ»ńē®µīćÕŹŚŌĆ£Content and Format of Investigational New Drug Applications (INDs) for Phase 1 Studies of Drugs, Including Well-Characterized, Therapeutic, Biotechnology-derived ProductŌĆØ�’╝ī�’╝īĶ»źµīćÕŹŚÕē¢µ×Éõ║å21 CFRõĖŁ312.22ÕÆī312.23Õģ│õ║ĵ£ĆÕłØĶ┐øÕģźńŠÄÕøĮõĖ┤Õ║ŖńĀöń®ČĶ»Ģķ¬īĶŹ»ńē®ńÜäµĢ░µŹ«Ķ”üµ▒é�’╝ī�’╝īÕģüĶ«ĖINDķśČµ«ĄµÅÉõ║żńÜäń¦Źń¦ŹńĀöń®ČńÜäµĢ░µŹ«ÕÅŖµĘ▒Õ║”�’╝ī�’╝īÕģʵ£ēµ×üÕż¦ńÜ䵌Āķ鬵Ʀ�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
2003Õ╣┤�’╝ī�’╝īFDAÕÅłÕ«ŻÕĖāõ║åµīćÕŹŚŌĆ£INDs for Phase 2 and Phase 3 StudiesŌĆØ�’╝ī�’╝īĶ»źµīćÕŹŚĶ┐øõĖƵŁźµÅÉõŠøńö│Ķ»Ęõ║║(INDķśČµ«Ą)Õģ│õ║ÄIIµ£¤/IIIµ£¤õĖ┤Õ║ŖÕī¢ÕŁ”ŃĆüńö¤õ║¦ŃĆüCMCõ┐Īµü»ńŁēÕåģÕ«╣ńÜäńøĖÕģ│Õ╗║Õ¬ŠÕÆīĶ”üµ▒é�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
2008Õ╣┤�’╝ī�’╝īFDAÕ«ŻÕĖāµīćÕŹŚŌĆ£CGMP for Phase 1 Investigational DrugsŌĆØ�’╝ī�’╝īĶ»źµīćÕŹŚĶ»”ń╗åÕÅÖĶ┐░õ║åŌģĀµ£¤õĖ┤Õ║ŖĶ»Ģķ¬īµĀĘÕōüńö¤õ║¦ĶÉĮÕ«×cGMPńÜäķŚ«ķóś�’╝ī�’╝īÕ╗║Ķ««µÄźń║│ĶŹ»ńē®Ķ┤©ķćÅµÄ¦ÕłČ(QC)ÕÄ¤ÕłÖõĮ£õĖ║cGMPńÜäõĖĆķā©Õłå�’╝ī�’╝īõ╗ÄĶĆīÕīģń«ĪŌģĀµ£¤õĖ┤Õ║ŖĶ»Ģķ¬īµĀĘÕōüńÜäĶ┤©ķćÅÕÆīµĖģķØֵƦ�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
2013Õ╣┤�’╝ī�’╝īFDAÕåŹµ¼ĪÕ«ŻÕĖāµīćÕŹŚŌĆ£Investigational New Drug Applications (INDs) - Determining Whether Human Research Studies Can Be Conducted Without an INDŌĆØ�’╝ī�’╝īĶ»źµīćÕŹŚµŚ©Õ£©ĶĄäÕŖ®õĖ┤Õ║Ŗńö│Ķ»Ęõ║║ńĪ«Õ«Üµ¢░ĶŹ»INDńö│Ķ»ĘõĖŗ�’╝ī�’╝īµČēÕÅŖńøĖÕģ│ńÜäńĀöń®Čµś»ÕÉ”Õ┐ģķ£ĆĶó½ńĀöń®Č�’╝ī�’╝īÕ”é21 CFR 312ķā©Õłå�’╝ø’╝ø�’╝øÕ╣ČĶ»”ń╗åĶ»┤µśÄµÖ░õĮĢµŚČķ£ĆĶ”üINDńö│Ķ»Ę�’╝ī�’╝īõĮĢń¦ŹµāģÕĮóõĖŹķ£ĆĶ”üINDńö│Ķ»Ę�’╝ī�’╝īńĪ«Õ«Üõ║åõĖĆÕ«ÜńÜäķĆéńö©Ķ¦äµ©Ī�ŃĆéŃĆéŃĆéŃĆé�ŃĆé

2. INDĶŹ»ÕŁ”ķā©Õłå-ÕłåÕØŚń╗åĶ┐░
ÕćŁĶ»üńŠÄÕøĮFDAÕ«ŻÕĖāńÜäńøĖÕģ│µīćÕŹŚńÜäĶ»”ń╗åÕåģÕ«╣�’╝ī�’╝īÕ£©ÕĘ«Õł½õĖ┤Õ║Ŗńö│µŖźķśČµ«Ą�’╝ī�’╝īÕģČÕ»╣ń½ŗÕ╝éĶŹ»ńē®-ĶŹ»ÕŁ”ķā©ÕłåńÜäĶ┤©µ¢ÖĶŹ»ŃĆüń©│Õø║µĆ¦ŃĆüĶ┤©ķćÅŃĆüÕłČÕēéńŁēÕåģÕ«╣�’╝ī�’╝īÕģʵ£ēĶ»”ń╗åńÜäÕłåµ«ĄńĀöń®ČĶ”üµ▒é�’╝ī�’╝īĶ»”µāģÕ”éõĖŗ’╝Ü
ŌśåĶ┤©µ¢ÖĶŹ»ķā©Õłå
ŌśåŌśåIµ£¤õĖ┤Õ║Ŗ
ÕłČÕżćÕĘźĶē║~µÅÉõŠøÕÉłµłÉÕĘźĶē║ńĀöń®ČńÜäń«ĆĶ”üµĆ╗ń╗ō�’╝ī�’╝īĶ»┤µśÄńÄ░µ£ēĶ»ĢÕłČĶ¦äµ©Ī�’╝ī�’╝īÕÉłµłÉĶ╣ŖÕŠäÕøŠõĖŁÕ╗║Ķ««µśÄńĪ«ÕÉäÕŖ×µ│ĢńÜäÕÅŹÕ║öµØĪõ╗ČŃĆüµēĆńö©Ķ»ĢÕēéŃĆüµ║ČÕēéŃĆüÕé¼Õī¢ÕēéńŁē�’╝ī�’╝īÕ╗║Ķ««µ£ĆÕģłÕģ│µ│©Õ»╣Ķ”üÕ«│ĶĄĘÕ¦ŗĶ┤©µ¢ÖńÜäĶ┤©ķćŵĢ░µŹ«ń¦»ń┤»�’╝ø’╝ø�’╝øÕøĀń▓ŠÕłČÕĘźĶē║ńÜäÕĘ«Õł½ÕÅ»ĶāĮÕĮ▒ÕōŹõ║¦ÕōüńÜäµØéĶ┤©Ķ░▒ŃĆüµÖČÕ×ŗŃĆüń▓ÆÕ║”ńŁē�’╝ī�’╝īķ£Ćµ│©ķćŹĶ»┤µśÄń▓ŚÕōüńÜäń║»Õī¢/ń▓ŠÕłČĶ”üķóå�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
ńē╣ÕŠüÕłżµ¢Ł~µŁżķśČµ«ĄµÅÉõŠøµö»µīüÕī¢ÕŁ”ń╗ōµ×äńÜäĶĄĘµ║ÉńĀöń®ČµĢ░µŹ«ÕŹ│ÕÅ»�’╝ø’╝ø�’╝øĶ»┤µśÄÕÅ»ĶāĮÕĮ▒ÕōŹµĖģķØֵƦńÜäńÉåÕī¢µĆ¦ÕŁÉ�’╝ī�’╝īÕ”éµČłĶ׏µĆ¦(ÕĘ«Õł½pHµ║ȵČ▓õĖŁ)ŃĆüń▓ÆÕ║”ŃĆüµÖČÕ×ŗńŁē�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕ╗║Ķ««Õ£©µŚ®µ£¤õĖ┤Õ║ŖķśČµ«ĄÕŹ│ńĪ«Õ«ÜĶŹ»ńö©µÖČÕ×ŗ�’╝ī�’╝īõĮåń▓ÆÕ║”Ķ┐śķ£ĆĶ”üĶ┐×ń│╗õĖ┤Õ║ŖńĀöń®ČńÜäµÄ©Ķ┐øõĖĆńø┤ń¦»ń┤»µĢ░µŹ«�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
ŌśåŌśåII/IIIµ£¤õĖ┤Õ║Ŗ
ÕłČÕżćÕĘźĶē║~µÅÉõ║żÕłČÕżćÕĘźĶē║ńÜäĶĮ¼ÕÅśÕÅŖńøĖÕģ│ńĀöń®ČĶĄäµ¢Ö�’╝ī�’╝īĶ»äõ╝░ÕÅśµŹóÕ»╣õ║¦ÕōüńÜäĶ┤©ķćÅÕÆīµĖģķØֵƦńÜäÕĮ▒ÕōŹ�’╝ø’╝ø�’╝øÕģ│õ║ÄÕīģń«Īõ║¦ÕōüµĖģķØֵƦńÜäńö¤õ║¦ÕŖ×µ│Ģ(Õ”éÕÅæķģĄõ║¦ÕōüńÜäń║»Õī¢ÕŖ×µ│Ģ)ńÜäÕÄåń©ŗµÄ¦ÕłČÕ║öµ£ēµĖģµÖ░ÕĮóĶ▓ī�’╝ø’╝ø�’╝øµÅÉõŠøĶĄĘÕ¦ŗĶ┤©µ¢ÖńÜäĶ┤©ķćÅµÄ¦ÕłČõ┐Īµü»(µ│ēµ║ÉŃĆüÕē¢µ×ÉĶ”üķóåŃĆüµŻĆµĄŗµĢłµ×£)�’╝ī�’╝īÕģ│õ║Äń╗ōµ×äķćŹÕż¦ńÜäĶ”üÕ«│ĶĄĘÕ¦ŗĶ┤©µ¢ÖÕ║öµÅÉõŠøĶ»”ń╗åńö¤õ║¦ÕĘźĶē║õ┐Īµü»�’╝ø’╝ø�’╝øµÅÉõŠøĶ”üÕ«│ÕŖ×µ│ĢŃĆüõĖŁÕ┐āõĮōńÜäµÄ¦ÕłČõ┐Īµü»�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
ńē╣ÕŠüÕłżµ¢Ł~µÅÉõŠøÕÉłńÉåµö»µīüĶŹ»ńē®Õī¢ÕŁ”ń╗ōµ×äńÜäĶ»üÕ«×�’╝ī�’╝īÕŹĢµÖČXĶĪŹÕ░äŃĆüµ×äĶ▒ĪÕē¢µ×ÉńŁēÕŻգ©IIIµ£¤µÅÉõŠø�’╝ø’╝ø�’╝øĶ┐×ń│╗õĖ┤Õ║ŖĶ»Ģķ¬īÕłČÕēéńÜäÕēéÕ×ŗńē╣ńé╣ÕÆīĶŹ»ńē®ńē╣ÕŠü�’╝ī�’╝īµÅÉõŠøĶ┐øõĖƵŁźÕ«īÕ¢äńÜäĶ┤©µ¢ÖĶŹ»ńÉåÕī¢µĆ¦ÕŁÉõ┐Īµü»�’╝ī�’╝īÕīģµŗ¼µČłĶ׏µĆ¦ŃĆüµÖČÕ×ŗŃĆüń▓ÆÕ║”ŃĆüµĖŚķĆÅµĆ¦ŃĆüµŚŗÕģēµĆ¦ŃĆüÕ╝Ģµ╣┐µĆ¦ŃĆüÕłåµ┤Šń│╗µĢ░ŃĆüńöĄń”╗ÕĖĖµĢ░ńŁē�’╝ī�’╝īÕģ│õ║ÄÕÅŻµ£ŹÕø║õĮōÕłČÕēé�’╝ī�’╝īÕ╗║Ķ««Õ░ĮµŚ®ńĀöń®ČÕģČĶ┤©µ¢ÖĶŹ»ńÜäµĖŚķĆÅµĆ¦�’╝ī�’╝īńøĖĶ»åÕģČBCSÕłåń▒╗�’╝ī�’╝īÕ»╣ÕłČÕēéÕżäµ¢╣ÕĘźĶē║Õ╝ĆÕÅæõ╗źÕÅŖõĮōÕż¢ķćŖµöŠĶ”üķóåńÜäÕ╗║Ķ«ŠÕŠłµ£ēĶĄäÕŖ®�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
Ōśåń©│Õø║µĆ¦ńĀöń®Č
ŌśåŌśåIµ£¤õĖ┤Õ║Ŗ
µÅÉõŠøÕĘ▓µ£ēńÜäń©│Õø║µĆ¦Ķ»Ģķ¬īµĢłµ×£ŃĆüÕÉÄń╗ŁńÜäń©│Õø║µĆ¦ńĀöń®ČÕ”äµā│�’╝ø’╝ø�’╝øÕģ│õ║ÄÕżŹµ║ČŃĆüń©ĆķćŖµł¢µĘʵĘåÕÉÄÕżÜµ¼ĪÕ║öńö©ńÜäÕłČÕēé�’╝ī�’╝īÕ║öÕ╝ĆÕ▒ĢõĮ┐ńö©õĖŁńÜäń©│Õø║µĆ¦ńĀöń®Č�’╝ø’╝ø�’╝øÕ╗║Ķ««Õ╝ĆÕ▒ĢÕĮ▒ÕōŹÕøĀń┤ĀńŁēĶ»Ģķ¬ī�’╝ī�’╝īõ╗źńøĖĶ»åĶŹ»ńē®ńÜäÕåģÕ£©ń©│Õø║µĆ¦µāģÕĮóŃĆüµĮ£Õ£©ńÜäķÖŹĶ¦ŻķĆöÕŠä�’╝ī�’╝īĶĄäÕŖ®ń©│Õø║µĆ¦Ķ»Ģķ¬īµØĪõ╗ČńÜäķĆēµŗ®ŃĆüÕē¢µ×ÉĶ”üķóåńÜäĶĆāÕ»¤�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕĘ▓µ£ēńÜäń©│Õø║µĆ¦ńĀöń®ČµĢłµ×£Õ║öµö»µīüµŗ¤õĖŠĶĪīńÜäõĖ┤Õ║ŖńĀöń®Č�’╝ī�’╝īÕīģń«ĪÕłČĶ«óõĖ┤Õ║ŖĶ»Ģķ¬īµŚČõ╗ŻĶŹ»ÕōüĶ┤©ķćÅńÜäń©│Õø║�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
ŌśåŌśåII/IIIµ£¤õĖ┤Õ║Ŗ
µĆ╗ń╗ōÕĘ▓ĶÄĘÕŠŚõ╗ŻĶĪ©µĆ¦µē╣µ¼ĪńÜäń©│Õø║µĆ¦Ķ»Ģķ¬īµĢ░µŹ«�’╝ø’╝ø�’╝øÕĮóĶ▓īĶ┤©µ¢ÖĶŹ»Õī¢ÕŁ”ÕÆīńē®ńÉåµĢÅµä¤µĆ¦�’╝ī�’╝īÕ”éÕģēµĢÅµä¤µĆ¦ŃĆüÕÉĖµ╣┐µĆ¦ńŁē�’╝ī�’╝īµĮ£Õ£©ńÜäķÖŹĶ¦ŻķĆöÕŠä�ŃĆéŃĆéŃĆéŃĆé�ŃĆéIŃĆüIIµ£¤õĖ┤Õ║ŖĶ»Ģķ¬īķĆÜÕĖĖÕ橵£¤ĶŠāķĢ┐�’╝ī�’╝īĶĆīµŗ¤ńö©õ║ÄõĖ┤Õ║ŖĶ»Ģķ¬īµĀĘÕōüńÜäń©│Õø║µĆ¦ĶĆāÕ»¤µŚČķŚ┤ÕŠłµ£ēķÖÉ�’╝ī�’╝īÕ╗║Ķ««ÕÅ»µÅÉõ║żńøĖÕģ│ńÜäµö»µīüµĆ¦ńĀöń®ČµĢ░µŹ«�’╝ī�’╝īõŠŗÕ”éõĖ┤Õ║ŖÕēŹµł¢µŚ®µ£¤õĖ┤Õ║ŖĶ»Ģķ¬īńÜäÕżäµ¢╣ŃĆüÕĘźĶē║ńøĖõ╝╝µē╣µ¼Īõ╗źÕÅŖµē╣ķćÅĶŠāÕ░ŵē╣µ¼ĪńŁēńÜäń©│Õø║µĆ¦ńĀöń®ČµĢłµ×£�ŃĆéŃĆéŃĆéŃĆé�ŃĆéĶ┐øÕģźIIIµ£¤õĖ┤Õ║ŖÕÉÄķĆÜÕĖĖÕ║öÕćŁĶ»üµīćÕ»╝ÕÄ¤ÕłÖĶ”üµ▒éÕ╝ĆÕ▒Ģń©│Õø║µĆ¦Ķ»Ģķ¬ī�’╝ī�’╝īõ╗źÕł®õŠ┐NDAńÜäńö│µŖź�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
ŌśåĶ┤©ķćÅķā©Õłå
ŌśåŌśåIµ£¤õĖ┤Õ║Ŗ
ÕłŚÕć║Ķ┤©ķćŵĀćÕćåńÜäķĪ╣ńø«ŃĆüĶ”üķóåÕÆīÕÅ»µÄźÕÅŚķÖÉÕ║”�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕ╗║Ķ««Õ»╣µČēÕÅŖµĖģķØֵƦńÜäµ£ēÕģ│ńē®Ķ┤©ŃĆüķüŚõ╝Āµ»ÆµĆ¦µØéĶ┤©ńŁēµŻĆµĄŗĶ”üķóåńÜäķĆéńö©µĆ¦õĖŠĶĪīĶĄĘµ║Éķ¬īĶ»ü�’╝ī�’╝īĶĄĘµ║ÉńĢīիܵØéĶ┤©Ķ░▒�’╝ø’╝ø�’╝øÕłČĶ«óķÖÉÕ║”Õ║öÕ¤║õ║ÄÕĘ▓µ£ēµē╣Õē¢µ×ɵĢ░µŹ«ńÜäń¦»ń┤»�’╝ī�’╝īõĖ┤Õ║ŖµĀĘÕōüńÜäµØéĶ┤©µ░┤Õ╣│õĖŹÕŠŚÕćīķ®ŠÕŖ©ńē®µĖģķØֵƦĶ»Ģķ¬īµĢ░µŹ«µēƵö»µīüńÜäÕōŹÕ║öµØéĶ┤©ńÜäµ░┤Õ╣│�’╝ø’╝ø�’╝øµÅÉõŠøÕĘ▓µ£ēµē╣µ¼Ī(Õ”éµĖģķØֵƦĶ»äõ╗ĘŃĆüń©│Õø║µĆ¦Ķ»Ģķ¬īńŁē)ÕÆīµŗ¤õĖŠĶĪīõĖ┤Õ║ŖĶ»Ģķ¬īµē╣µ¼Ī(Ķŗźµ£ē)ńÜäµē╣Õē¢µ×ɵĢ░µŹ«�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
ŌśåŌśåII/IIIµ£¤õĖ┤Õ║Ŗ
µÅÉõŠøÕē¢µ×ÉĶ”üķóåķā©Õłåķ¬īĶ»üµĢłµ×£µæśĶ”ü(ÕÅ»ÕłŚĶĪ©�’╝ī�’╝īÕ”éõĖōÕ▒׵ƦŃĆüń╗åÕ»åÕ║”ŃĆüÕćåńĪ«Õ║”ŃĆüń║┐µĆ¦ŃĆüÕ«ÜķćÅķÖÉ/µŻĆµĄŗķÖÉńŁē)�’╝ø’╝ø�’╝øń╗¦ń╗ŁõĖŠĶĪīµØéĶ┤©Ķ░▒ńÜäÕłżµ¢Ł�’╝ø’╝ø�’╝øÕ»╣Ķ»üµ¢ÖĶŹ»ÕÉłµłÉÕĘźĶē║ÕÅśµŹóńłåÕÅæńÜäµ¢░µØéĶ┤©ÕÆīÕłČÕēéõĖŁµ¢░ÕÅæµśÄńÜäķÖŹĶ¦Żõ║¦ÕōüõĖŠĶĪīÕ«ÜµĆ¦ÕÆīÕ«ÜķćÅńĀöń®Č�’╝ī�’╝īÕ╗║Ķ««ńö│µŖźIŃĆüIIµ£¤õĖ┤Õ║ŖµŚČńĪ«Õ«ÜĶ┤©µ¢ÖĶŹ»õĖ╗Ķ”üµØéĶ┤©õ╗źÕÅŖÕłČÕēéńÜäõĖ╗Ķ”üķÖŹĶ¦Żõ║¦Õōü�’╝ø’╝ø�’╝øķ揵¢░Ķ»äõ╝░ÕģłÕēŹIµ£¤µł¢IIµ£¤ńÜäĶ┤©ķćŵĀćÕćåÕÆīÕÅ»µÄźÕÅŚķÖÉÕ║”�’╝ī�’╝īÕćŁĶ»üńø«õ╗ŖńÜäńĀöń®ČķśČµ«ĄĶ┐øõĖƵŁźĶ»äõ╝░ÕÆīĶ░āĶ¦Ż�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕģ│õ║ÄķÜŠµ║ȵƦÕÅŻµ£ŹÕø║õĮōÕłČÕēé�’╝ī�’╝īÕ╗║Ķ««ń¦»ń┤»ÕłČÕēéµēĆńö©Ķ┤©µ¢ÖĶŹ»ńÜäń▓ÆÕ║”µ╝½ĶĪŹµĢ░µŹ«�’╝ī�’╝īÕ╗║Ķ«ŠĶŹ»ńē®Õ╝ĆÕÅæµŚ®µ£¤ŃĆüÕÉĵ£¤ĶÄĘÕŠŚµĢ░µŹ«õĖÄõĮōÕåģń¢ŚµĢłńÜäńøĖÕģ│µĆ¦�’╝ø’╝ø�’╝øÕ╗║Ķ«Šµ║ČÕć║Õ║”/ķćŖµöŠÕ║”Ķ”üķóå�’╝ī�’╝īĶ┐×ń│╗ĶŹ»ńē®ńē╣ÕŠüķĆēµŗ®õ╗ŗĶ┤©ÕÆīĶ»Ģķ¬īĶ”üķóå�’╝ī�’╝īÕ╗║Ķ««Õ»╣õĖ┤Õ║ŖÕēŹĶ»Ģķ¬īµĀĘÕōüŃĆüÕÉäµ£¤õĖ┤Õ║ŖĶ»Ģķ¬īµĀĘÕōüŃĆüń©│Õø║µĆ¦Ķ»Ģķ¬īµĀĘÕōüńÜäµ║ČÕć║/ķćŖµöŠĶĪīõĖ║õĖŠĶĪīĶĆāÕ»¤�’╝ī�’╝īÕ╗║Ķ«ŠĶŹ»ńē®Õ╝ĆÕÅæµŚ®µ£¤ŃĆüÕÉĵ£¤ĶÄĘÕŠŚµĢ░µŹ«õĖÄõĮōÕåģń¢ŚµĢłńÜäńøĖÕģ│µĆ¦�ŃĆéŃĆéŃĆéŃĆé�ŃĆéµÅÉõ║żÕÉäķĪ╣õĖ┤Õ║ŖĶ»Ģķ¬īµĀĘÕōüńÜäµē╣Õē¢µ×ɵĢ░µŹ«�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
ŌśåÕłČÕēéķā©Õłå
ŌśåŌśåIµ£¤õĖ┤Õ║Ŗ
ķĆÜÕĖĖµÄźń║│ńÜäÕēéÕ×ŗĶŠāķćÅń«Ćµ£┤�’╝ī�’╝īõŠŗÕ”éÕÅŻµ£ŹÕłČÕēéµÄźń║│ń▓ēµ£½ĶŻģĶāČÕøŖ�’╝ī�’╝īµł¢ĶĆģÕłČÕżćµłÉµĘĘµé¼µČ▓ŃĆüµ║ȵČ▓ńŁē�’╝ī�’╝īõ╗źÕł®õŠ┐ÕēéķćŵÄóń┤ó�’╝ī�’╝īµŁżķśČµ«ĄńÜäÕēéÕ×ŗÕÆīÕżäµ¢╣ÕĘźĶē║Ķ┐śõ┐ØÕŁśÕŠłÕż¦ńÜäõĖŹńĪ«Õ«ÜµĆ¦�’╝ī�’╝īõĖŹµś»ĶŹ»ÕŁ”Ķ»äõ╗ĘńÜäķćŹńé╣�’╝ī�’╝īķćŹńé╣µś»Õīģń«ĪõĖ┤Õ║ŖĶ»Ģķ¬īµĀĘÕōüńÜäń©│Õø║ŃĆüµĖģķØÖ�ŃĆéŃĆéŃĆéŃĆé�ŃĆéõĮåÕģ│õ║ĵŚĀĶÅīÕłČÕēé�’╝ī�’╝īÕć║õ║ĵĖģķØֵƦńÜäµĆØķćÅ�’╝ī�’╝īÕ║öµÅÉõŠøĶ»”ń╗åńÜäńüŁĶÅī/ķÖżĶÅīÕĘźĶē║µØĪõ╗Č�’╝ī�’╝īÕłČÕżćÕĘźĶē║Õ║öĶāĮÕīģń«Īõ║¦ÕōüńÜ䵌ĀĶÅī�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕ║öµ│©ķćŹĶ»┤µśÄõĖ┤Õ║ŖĶ»Ģķ¬īµŗ¤ńö©ÕłČÕēéÕÆīµ»ÆńÉåÕŁ”Ķ»Ģķ¬īµēĆńö©ÕłČÕēéÕ£©ńö¤õ║¦ŃĆüńē╣ÕŠüµ¢╣ķØóńÜäÕĘ«Õł½�’╝ī�’╝īĶ«©Ķ«║Ķ┐Öõ║øÕĘ«Õł½Õ»╣µĖģķØֵƦÕÅ»ĶāĮńÜäÕĮ▒ÕōŹµ░┤Õ╣│�’╝ī�’╝īµĆ╗õ╣ŗ�’╝ī�’╝īĶ”üÕīģń«Īńö©õ║ÄõĖ┤Õ║ŖÕēŹÕŖ©ńē®Ķ»Ģķ¬īŃĆüõĖ┤Õ║ŖĶ»Ģķ¬īńŁēµēĆńö©ĶŹ»ńē®ńÜäĶ┤©ķćÅÕģʵ£ēÕÅ»µ»öµĆ¦�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕł½ńÜäĶ”üĶ»┤µśÄĶ┤©µ¢ÖÕÆīÕłČÕēéńÜäÕłČÕżćÕÄåń©ŗµś»ÕÉ”µśŠńż║Õć║õ╗╗õĮĢµĮ£Õ£©ńÜäõ║║õĮōÕŹ▒Õ«│õ┐ĪÕÅĘ�’╝ī�’╝īĶŗźµ£ē�’╝ī�’╝īÕ║öÕ»╣Ķ┐Öõ║øµĮ£Õ£©ńÜäÕŹ▒ķÖ®õ┐ĪÕÅĘõĖŠĶĪīÕē¢µ×É�’╝ī�’╝īÕÅÖĶ┐░ńøæµĄŗÕ”äµā│�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
ŌśåŌśåII/IIIµ£¤õĖ┤Õ║Ŗ
µÅÉõ║żIµ£¤µł¢IIµ£¤õĖ┤Õ║ŖµŚČõ╗ŻÕēéÕ×ŗŃĆüÕżäµ¢╣ŃĆüÕĘźĶē║ńÜäĶĮ¼ÕÅśÕÅŖńøĖÕģ│ńĀöń®ČĶĄäµ¢Ö�’╝ī�’╝īµ║ČÕć║ĶĪīõĖ║ńŁēĶ┤©ķćÅńē╣ÕŠüÕÅ»ĶāĮÕģʵ£ēµĮ£Õ£©õĖ┤Õ║ŖńøĖÕģ│µĆ¦�’╝ī�’╝īĶ»ĘÕģ│µ│©ÕÅśµŹóÕ»╣Ķ┐Öõ║øĶ┤©ķćÅńē╣ÕŠüńÜäÕĮ▒ÕōŹ�’╝ī�’╝īĶ»äõ╗ʵŚ®µ£¤õĖ┤Õ║ŖĶ»Ģķ¬īÕłČÕēéõĖÄÕÉÄń╗Łµŗ¤õĮ┐ńö©ÕłČÕēéńÜäńøĖÕģ│µĆ¦�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕģ│õ║ÄIŃĆüIIµ£¤ńö│µŖź�’╝ī�’╝īÕ”éÕĘ▓µśÄńĪ«Ķ”üÕ«│ńö¤õ║¦ÕŖ×µ│Ģ�’╝ī�’╝īÕ║öń║¬ÕĮĢĶ”üÕ«│ÕŖ×µ│ĢńÜäµÄ¦ÕłČÕÆīõĖŁÕ┐āõĮōńÜäµÄ¦ÕłČõ┐Īµü»�ŃĆéŃĆéŃĆéŃĆé�ŃĆéIIIµ£¤õĖ┤Õ║ŖĶ»Ģķ¬īµś»ńĪ«Ķ«żĶŹ»ÕōüµĖģķØֵƦµ£ēńö©µĆ¦µ£ĆõĖ╗Ķ”üńÜäĶ»Ģķ¬īķā©Õłå�’╝ī�’╝īIŃĆüIIµ£¤õĖ┤Õ║ŖĶ»Ģķ¬īµēĆńö©ńÜäµĀĘÕōüµś»Õģ│ĶüöĶŹ»ÕōüńÜäµĖģķØÖµ£ēńö©µĆ¦õĖÄõ║¦ÕōüĶ┤©ķćÅÕ▒׵ƦńÜäĶ”üÕ«│µē╣µ¼Ī�’╝ī�’╝īÕģ│õ║ĵ£¬µØźµ¢░ĶŹ»õĖŖÕĖéńö│Ķ»Ę(NDA)ńö│µŖźµŚČÕłČĶ«óÕæ©Õģ©ńÜäĶ┤©ķćÅµÄ¦ÕłČń│╗ń╗¤ÕŠłµś»õĖ╗Ķ”ü�’╝ī�’╝īÕ╗║Ķ««ķ½śÕ║”Õģ│µ│©IŃĆüIIµ£¤õĖ┤Õ║ŖĶ»Ģķ¬īµĀĘÕōüńÜäCMCńøĖÕģ│õ┐Īµü»�ŃĆéŃĆéŃĆéŃĆé�ŃĆéķĆÜÕĖĖµś»ÕćŁĶ»üÕłČĶ«óÕĢåõĖÜÕī¢ńö¤õ║¦µØźÕ»╣ IŃĆüIIµ£¤õĖ┤Õ║ŖµĀĘÕōüńÜäńö¤õ║¦ÕÆīÕģČõ╗¢ĶŹ»ÕŁ”ńĀöń®Čõ║ŗµāģõĖŠĶĪīÕÉłńÉåÕ«ēµÄÆ�’╝ī�’╝īÕŬń«Īķś╗µŁóNDAõ╣ŗÕēŹÕåŹńłåÕÅæÕĮ▒ÕōŹõ║¦ÕōüĶ┤©ķćÅńÜäķćŹÕż¦ÕÅśµŹó�’╝ī�’╝īÕó×Õ╝║Õ»╣ÕĘźĶē║µÄ¦ÕłČõ┐Īµü»ŃĆüĶ”üÕ«│Ķ┤©ķćÅõ┐Īµü»ńÜäńĮæń╗£�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
õĖŁÕøĮCFDA&ń½ŗÕ╝éĶŹ»INDķśČµ«Ą-ĶŹ»ÕŁ”ńĀöń®Č
1. CFDAÕģ│õ║Äń½ŗÕ╝éĶŹ»INDķśČµ«ĄÕ«ĪĶ»äńÜäńøĖÕģ│µö┐ńŁ¢
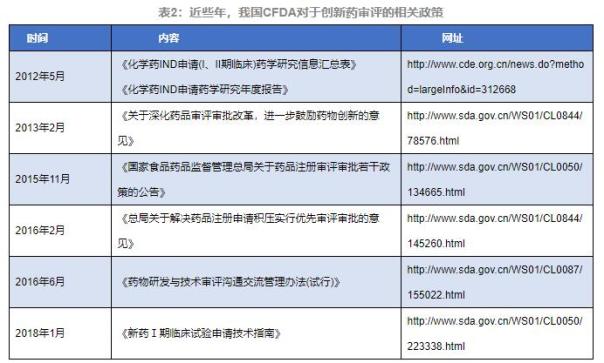
2012Õ╣┤5µ£ł�’╝ī�’╝īÕłČĶ«óŃĆŖÕī¢ÕŁ”ĶŹ»INDńö│Ķ»Ę(IŃĆüIIµ£¤õĖ┤Õ║Ŗ)ĶŹ»ÕŁ”ńĀöń®Čõ┐Īµü»µ▒ćµĆ╗ĶĪ©ŃĆŗŃĆüŃĆŖÕī¢ÕŁ”ĶŹ»INDńö│Ķ»ĘĶŹ»ÕŁ”ńĀöń®ČÕ╣┤Õ║”µŖźÕæŖŃĆŗ�’╝ī�’╝īńø«ńÜäõĖ║Õł®õŠ┐ń½ŗÕ╝éĶŹ»ńÜäĶŹ»ÕŁ”Õ«ĪĶ»äõ╗źÕÅŖõĖ┤Õ║ŖĶ»Ģķ¬īµŚČõ╗ŻÕÉÄń╗ŁĶŹ»ÕŁ”ńĀöń®Čõ┐Īµü»ńÜäĶĮ¼ÕŖ©µÅÉõ║ż�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
2013Õ╣┤2µ£ł�’╝ī�’╝īÕ«ŻÕĖāŃĆŖÕģ│õ║ĵĘ▒Õī¢ĶŹ»ÕōüÕ«ĪĶ»äÕ«Īµē╣ÕłĘµ¢░�’╝ī�’╝īĶ┐øõĖƵŁźÕŗēÕŖ▒ĶŹ»ńē®ń½ŗÕ╝éńÜäµäÅĶ¦üŃĆŗ(ÕøĮķŻ¤ĶŹ»ńøæµ│©[2013]37ÕÅĘ)�’╝ī�’╝īµśÄńĪ«õ║åÕŗēÕŖ▒õ╗źõĖ┤Õ║Ŗõ╗ĘÕĆ╝õĖ║Õ»╝ÕÉæńÜäĶŹ»ńē®ń½ŗÕ╝éŃĆüĶ░āĶ¦Żń½ŗÕ╝éĶŹ»ńē®õĖ┤Õ║ŖĶ»Ģķ¬īńö│Ķ»ĘńÜäÕ«ĪĶ»äµłśńĢźŃĆüõ╝śÕī¢ń½ŗÕ╝éĶŹ»ńē®Õ«ĪĶ»äµĄüń©ŗ�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
2015Õ╣┤11µ£ł�’╝ī�’╝īÕ«ŻÕĖāŃĆŖÕøĮÕ«ČķŻ¤ńē®ĶŹ»ÕōüńøæĶ¦åµ▓╗ńÉåµĆ╗Õ▒ĆÕģ│õ║ÄĶŹ»Õōüµ│©ÕåīÕ«ĪĶ»äÕ«Īµē╣ĶŗźÕ╣▓µö┐ńŁ¢ńÜäķĆÜÕæŖŃĆŗ(2015Õ╣┤ń¼¼230ÕÅĘ)�’╝ī�’╝īÕ╝║Ķ░āÕ£©Iµ£¤ŃĆüIIµ£¤õĖ┤Õ║ŖĶ»Ģķ¬īÕ«īµłÉÕÉÄ�’╝ī�’╝īńö│Ķ»Ęõ║║Õ║öÕ«×µŚČµÅÉõ║żĶ»Ģķ¬īµĢłµ×£ÕÅŖõĖŗõĖƵ£¤õĖ┤Õ║ŖĶ»Ģķ¬īĶ«ĪÕłÆ�ŃĆéŃĆéŃĆéŃĆé�ŃĆéµ£¬ÕÅæµśÄµĖģķØֵƦķŚ«ķóśńÜä�’╝ī�’╝īÕŻգ©õĖÄĶŹ»Õ«ĪõĖŁÕ┐āńøĖÕÉīÕÉÄĶĮ¼ÕģźõĖŗõĖƵ£¤õĖ┤Õ║ŖĶ»Ģķ¬ī�ŃĆéŃĆéŃĆéŃĆé�ŃĆéII/IIIµ£¤õĖ┤Õ║ŖĶ»Ģķ¬īĶÖĮõĖŹÕåŹķ£ĆĶ”üÕ«Īµē╣�’╝ī�’╝īõĮåõ╗Źķ£Ćńö│Ķ»Ęõ║║ÕćŁĶ»üń½ŗÕ╝éĶŹ»ńÜäńĀöÕÅæń║¬ÕŠŗÕÉłńÉåÕłČĶ«óĶŹ»ÕŁ”ńĀöÕÅæµłśńĢź�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
2016Õ╣┤6µ£ł�’╝ī�’╝īµĆ╗Õ▒ĆÕ«ŻÕĖāŃĆŖĶŹ»ńē®ńĀöÕÅæõĖĵēŗĶē║Õ«ĪĶ»äńøĖÕÉīõ║żµĄüµ▓╗ńÉåµŁźõ╝É(Ķ»ĢĶĪī)ŃĆŗ�’╝ī�’╝īÕģČńø«ńÜ䵜»Õ╗║Ķ««ńö│Ķ»Ęõ║║ķćŹĶ¦åIIIõĖ┤Õ║ŖÕēŹńÜäĶŹ»ÕŁ”ńøĖÕÉīõ║żµĄüõ╝Ü�’╝ī�’╝īÕ╣ČÕģģÕłåĶ«©Ķ«║IIIµ£¤õĖ┤Õ║ŖµĀĘÕōüńÜäńö¤õ║¦Ķ”üµ▒éÕÅŖÕÉÄń╗ŁńÜäĶŹ»ÕŁ”ńĀöÕÅæÕ”äµā│�’╝ī�’╝īõ║æõ║æÕ░åµ£ēÕŖ®õ║Äńö│Ķ»Ęõ║║Õ£©Ķ”üÕ«│ńÜäIIIõĖ┤Õ║ŖĶ»Ģķ¬īõĖŁĶÄĘÕÅ¢ÕģģÕłåńÜäµö»µīüNDAńÜäCMCµĢ░µŹ«�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
2018Õ╣┤1µ£ł�’╝ī�’╝īÕ«ŻÕĖāŃĆŖµ¢░ĶŹ»Iµ£¤õĖ┤Õ║ŖĶ»Ģķ¬īńö│Ķ»ĘµēŗĶē║µīćÕŹŚŃĆŗ�’╝ī�’╝īńø«ńÜ䵜»µśÄńĪ«µ¢░ĶŹ»Iµ£¤õĖ┤Õ║ŖĶ»Ģķ¬īńÜäµēŗĶē║Ķ”üµ▒é�’╝ī�’╝īµÅÉķ½śIµ£¤õĖ┤Õ║ŖĶ»Ģķ¬īńö│µŖźĶĄäµ¢ÖńÜäĶ┤©ķćÅ�’╝ø’╝ø�’╝øķĆÜĶ┐ćĶ¦äĶīāIµ£¤õĖ┤Õ║ŖĶ»Ģķ¬īĶĄäµ¢ÖńÜäµĢ░µŹ«Ķ”üµ▒é�’╝ī�’╝īń╝®ń¤Łµ¢░ĶŹ»ńĀöÕÅæÕ橵£¤�’╝ī�’╝īÕŖĀķƤµ¢░ĶŹ»õĖŖÕĖéÕÄåń©ŗ�’╝ø’╝ø�’╝øĶĄäÕŖ®µ¢░ĶŹ»µ│©Õåīńö│Ķ»Ęõ║║ńö│Ķ»ĘIµ£¤õĖ┤Õ║ŖĶ»Ģķ¬ī�’╝ī�’╝īµÅÉķ½śµ¢░ĶŹ»ńĀöÕÅæõĖÄÕ«ĪĶ»äµĢłńÄć�’╝ī�’╝ī�’╝ø’╝ø�’╝øŃü¬ĶŗüÕōēÕÆöÕ»░ńü┐Ķé┤ŃäÆ�’╝ī�’╝īÕīģń«ĪõĖ┤Õ║ŖĶ»Ģķ¬īĶ┤©ķćÅ�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
2. µ£Ćµ¢░ńēłŃĆŖµ¢░ĶŹ»Iµ£¤õĖ┤Õ║ŖĶ»Ģķ¬īńö│Ķ»ĘµēŗĶē║µīćÕŹŚŃĆŗ~ĶŹ»ÕŁ”ńĀöń®ČńøĖÕģ│ÕåģÕ«╣
2018Õ╣┤1µ£ł�’╝ī�’╝īµĆ╗Õ▒ĆÕ«ŻÕĖāõ║åŃĆŖµ¢░ĶŹ»ŌģĀµ£¤õĖ┤Õ║ŖĶ»Ģķ¬īńö│Ķ»ĘµēŗĶē║µīćÕŹŚŃĆŗ�’╝ī�’╝īÕ»╣ńö│Ķ»ĘŌģĀµ£¤õĖ┤Õ║ŖńĀöń®ČńÜäÕī¢ÕŁ”ĶŹ»Õōü�’╝ī�’╝īķ£ĆĶ”üµÅÉõŠøõĖŗÕłŚĶŹ»ÕŁ”ńĀöń®ČĶĄäµ¢Ö�’╝ī�’╝īÕÉīµŚČ�’╝ī�’╝īÕćŁĶ»üķÖäõ╗ČĶĪ©µĀ╝µĆ╗ń╗ōµĢ┤ńÉåÕÆīµÅÉõŠøÕī¢ÕŁ”ĶŹ»ÕōüŌģĀµ£¤õĖ┤Õ║ŖĶ»Ģķ¬īńö│Ķ»ĘĶŹ»ÕŁ”ńĀöń®Čõ┐Īµü»µ▒ćµĆ╗ĶĪ©Õ╣ČńöĄÕŁÉµÅÉõ║ż�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
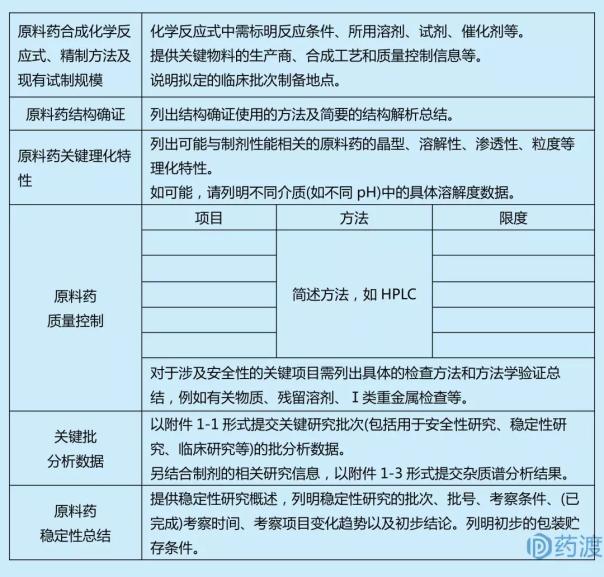
ŌśåĶ┤©µ¢ÖĶŹ»õ┐Īµü»
ŌśåŌśåńö¤õ║¦ÕÄéÕĢå
Õ║öķĆÆõ║żĶ┤©µ¢ÖĶŹ»ńö¤õ║¦ÕÄéÕĢå(Õīģµŗ¼ńö¤õ║¦ŃĆüńŻ©ń╗ā)ńÜäÕ«īµĢ┤Õ£░ńé╣�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
ŌśåŌśåÕłČÕżćÕĘźĶē║
Õ║öµÅÉõŠøĶ┤©µ¢ÖĶŹ»ÕłČÕżćÕĘźĶē║ĶĄäµ¢Ö�’╝ī�’╝īÕīģµŗ¼ÕÅŹÕ║öµĄüń©ŗÕøŠ�’╝ī�’╝īµ│©µśÄÕĘźĶē║õĖŁõĮ┐ńö©ńÜäĶ»ĢÕēéŃĆüµ║ČÕēéÕÆīÕé¼Õī¢ÕēéńŁē�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕģ│õ║ĵğń║│ÕÅæķģĄÕĘźĶē║ŃĆüµÅÉÕÅ¢ÕĘźĶē║ÕłČÕżćõ╗źÕÅŖÕżÜĶéĮŃĆüÕ░ÅÕłåÕŁÉµĀĖķģĖĶŹ»ńē®ńŁē�’╝ī�’╝īķ£ĆĶ”üµÅÉõŠøµø┤ÕżÜńÜäÕłČÕżćÕĘźĶē║õ┐Īµü»�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕģ│õ║ĵŚĀĶÅīĶ┤©µ¢ÖĶŹ»ķ£ĆµÅÉõŠøńüŁĶÅī/ķÖżĶÅīÕĘźĶē║ÕÆīµŚĀĶÅīÕīģń«ĪµŁźõ╝É�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
ŌśåŌśåń╗ōµ×äńĪ«Ķ»ü
Õ║öµÅÉõŠøń╗ōµ×äńĪ«Ķ»üõĮ┐ńö©ńÜäĶ”üķóåŃĆüÕøŠĶ░▒ÕÅŖń«ĆĶ”üńÜäń╗ōµ×äÕē¢µ×ɵĆ╗ń╗ō�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
ŌśåŌśåńÉåÕī¢µĆ¦ÕŁÉ
Õ║öÕłŚÕć║ÕĘ▓ńĀöń®ČńÜäÕÅ»ĶāĮõĖÄÕłČÕēéµĆ¦ĶāĮńøĖÕģ│ńÜäĶ┤©µ¢ÖĶŹ»ńÜäµÖČÕ×ŗŃĆüµČłĶ׏Õ║”ŃĆüµĖŚķĆÅµĆ¦ŃĆüń▓ÆÕ║”ńŁēĶ”üÕ«│ńÉåÕī¢ńē╣ÕŠü�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕ”éÕÅ»ĶāĮ�’╝ī�’╝īÕłŚµśÄÕĘ«Õł½õ╗ŗĶ┤©(Õ”éÕĘ«Õł½pH)õĖŁńÜäĶ»”ń╗åµČłĶ׏Õ║”µĢ░µŹ«�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
ŌśåŌśåĶ┤©ķćÅµÄ¦ÕłČ
Õ║öµÅÉõŠøĶĄĘµ║ÉńÜäĶ┤©ķćŵĀćÕćå�’╝ī�’╝īĶ»┤µśÄµŻĆµ¤źķĪ╣ńø«ŃĆüÕÅ»µÄźÕÅŚńÜäķÖÉÕ║”ŃĆüÕē¢µ×ÉĶ”üķóå�’╝ī�’╝īµÅÉõŠøõ╗ŻĶĪ©µĆ¦ÕøŠĶ░▒�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕ£©ĶŹ»ÕōüÕ╝ĆÕÅæÕłØµ£¤�’╝ī�’╝īõĖŹķ£ĆĶ”üµÅÉõ║żÕæ©Õģ©Õ«īµĢ┤ńÜäÕē¢µ×ÉĶ”üķóåķ¬īĶ»üĶĄäµ¢Ö�’╝ī�’╝īõĮåĶć│Õ░æÕ║öµÅÉõŠøĶ”üķóåńÜäõĖōÕ▒׵ƦŃĆüĶ┐ģķƤÕ║”ńŁēĶ”üÕ«│ķ¬īĶ»üõ┐Īµü»�ŃĆéŃĆéŃĆéŃĆé�ŃĆé Õ║öµÅÉõŠøµĀĘÕōüńŻ©ń╗āµŖźÕæŖõ╣”�ŃĆéŃĆéŃĆéŃĆé�ŃĆéµÅÉõŠøĶ”üÕ«│ńĀöń®Čµē╣µ¼Ī(Õ”éńö©õ║ĵĖģķØֵƦńĀöń®ČŃĆüń©│Õø║µĆ¦ńĀöń®ČŃĆüõĖ┤Õ║ŖńĀöń®ČńŁē)ńÜäµē╣Õē¢µ×ɵĢ░µŹ«�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕ║öµÅÉõŠøĶĄĘµ║ÉńÜäµØéĶ┤©Ķ░▒Õē¢µ×ɵĢłµ×£ŃĆüµĮ£Õ£©ķüŚõ╝Āµ»ÆµĆ¦µØéĶ┤©µÄ¦ÕłČµłśńĢźÕÆīÕē¢µ×Éõ┐Īµü»�ŃĆéŃĆéŃĆéŃĆé��’╝¤�’╝¤�’╝¤�’╝¤�’╝¤Õł╣µÅĪĶ¬īCH M7µīćÕŹŚńĀöń®ČÕ╣ȵÅÉõ║żµŖźÕæŖ�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
ŌśåŌśåń©│Õø║µĆ¦
Õ║öµÅÉõŠøĶ┤©µ¢ÖĶŹ»ń©│Õø║µĆ¦ńĀöń®ČĶĄäµ¢Ö�’╝ī�’╝īÕłŚµśÄµÄźń║│ńÜäÕē¢µ×ÉĶ”üķóå�’╝ī�’╝īÕÅ»ńö©ÕłŚĶĪ©ÕĮóÕ╝ÅķĆÆõ║żµÄźĶĪ©µĆ¦µĀĘÕōüńÜäĶĄĘµ║ɵĢ░µŹ«ÕÅŖÕģČõ╗¢µö»µīüµĆ¦ń©│Õø║µĆ¦ńĀöń®ČµĢ░µŹ«�’╝ī�’╝īÕ║öµÅÉõŠøĶ”üÕ«│ķĪ╣ńø«ńÜäõ╗ŻĶĪ©µĆ¦ÕøŠĶ░▒�ŃĆéŃĆéŃĆéŃĆé�ŃĆéń©│Õø║µĆ¦µĢ░µŹ«Õ║öĶāĮµö»µīüµ¢░ĶŹ»ńÜäńÉåÕī¢ÕÅéµĢ░Õ£©Õ”äµā│ńÜäõĖ┤Õ║ŖńĀöń®ČµŚČõ╗ŻÕłćÕÉłĶ”üµ▒é�’╝ī�’╝īĶŗźµś»Õ”äµā│ńÜäĶ»Ģķ¬īÕ橵£¤µ×üń¤Ł�’╝ī�’╝īÕÅ»µÅÉõŠøµ£ēķÖÉńÜäµö»µīüµĆ¦ń©│Õø║µĆ¦µĢ░µŹ«�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕ£©ńĪ«õ┐ØõĖ┤Õ║ŖĶ»Ģķ¬īµŚČõ╗ŻĶŹ»ńē®ńÜäń©│Õø║µĆ¦ńÜäÕ¤║ńĪĆõĖŖ�’╝ī�’╝īķĆɵŁźń¦»ń┤»ń©│Õø║µĆ¦µĢ░µŹ«�’╝ī�’╝īµö»µīüĶ┐øõĖƵŁźńÜäõĖ┤Õ║ŖÕ╝ĆÕÅæ�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
ŌśåŌśåÕīģĶŻģÕÅŖĶ┤«ÕŁś
Õ║öÕłŚµśÄńÜäńø┤µÄźµÄźĶ¦”ÕīģĶŻģĶ┤©µ¢ÖÕÅŖĶ┤«ÕŁśµØĪõ╗Č�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
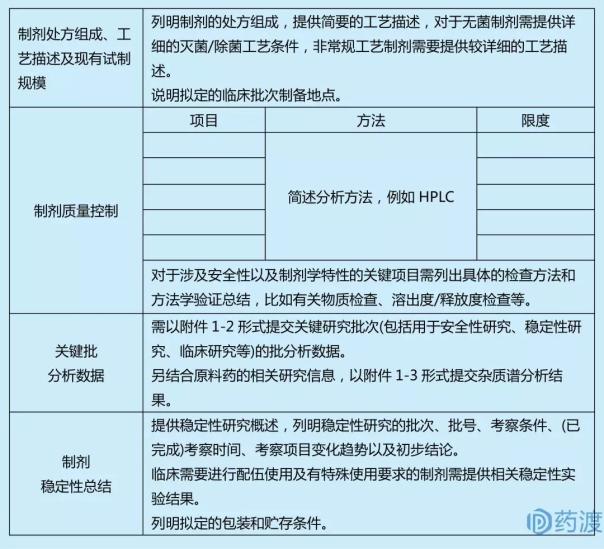
ŌśåÕłČÕēéõ┐Īµü»
ŌśåŌśåÕēéÕ×ŗÕÅŖõ║¦Õōüń╗䵳É
Õ║öÕłŚĶĪ©Ķ»┤µśÄÕłČÕēéńÜäÕżäµ¢╣ń╗䵳ÉÕÅŖńö©ķćÅ�’╝ī�’╝īÕģ│õ║ÄÕłČÕēéÕĘźĶē║õĖŁõĮ┐ńö©õĮåµ£Ćń╗łÕÄ╗ķÖżńÜäń╗äÕłåõ╣¤Õ║öÕłŚÕć║�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕłČÕēéõĖŁńÜäĶŠģµ¢ÖÕ║öÕłćÕÉłĶŹ»ńö©Ķ”üµ▒é�’╝ø’╝ø�’╝øÕģ│õ║ĵĄĘÕåģÕż¢ÕłČÕēéõĖŁÕ░ܵ£¬õĮ┐ńö©Ķ┐ćńÜäÕģ©µ¢░ĶŠģµ¢Ö�’╝ī�’╝īÕ║öõĖŠĶĪīÕģ│Ķüöńö│µŖź�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
ŌśåŌśåńö¤õ║¦ÕÄéÕĢåÕÉŹń¦░õĖÄÕ£░ńé╣
Õ║öķĆÆõ║żõĖ┤Õ║ŖĶ»Ģķ¬īńö©ÕłČÕēéńö¤õ║¦ÕÄéÕĢå(Õīģµŗ¼ńö¤õ║¦ŃĆüÕīģĶŻģŃĆüńŻ©ń╗ā)ńÜäÕ«īµĢ┤Õ£░ńé╣�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
ŌśåŌśåńö¤õ║¦ÕĘźĶē║ÕÆīÕĘźĶē║µÄ¦ÕłČ
Õ║öµÅÉõŠøńö¤õ║¦ÕĘźĶē║õ┐Īµü»�’╝ī�’╝īÕīģµŗ¼ÕĘźĶē║µĄüń©ŗÕøŠ�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕģ│õ║ĵŚĀĶÅīÕłČÕēéÕ║öµÅÉõŠøńüŁĶÅīÕĘźĶē║ÕÆīµŚĀĶÅīÕīģń«ĪµŁźõ╝É�’╝ø’╝ø�’╝øķØ×ķĆÜõŠŗÕĘźĶē║ÕłČÕēéÕ║öµÅÉõŠøĶŠāĶ»”ń╗åńÜäÕĘźĶē║ÕĮóĶ▓ī�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
ŌśåŌśåĶ┤©ķćÅµÄ¦ÕłČ
Õ║öµÅÉõŠøĶĄĘµ║ÉńÜäĶ┤©ķćŵĀćÕćå�ŃĆéŃĆéŃĆéŃĆé�ŃĆéĶ»┤µśÄµŻĆµ¤źķĪ╣ńø«ÕÅ»µÄźÕÅŚńÜäķÖÉÕ║”ŃĆüÕē¢µ×ÉĶ”üķóåŃĆüõ╗ŻĶĪ©µĆ¦ÕøŠĶ░▒�ŃĆéŃĆéŃĆéŃĆé�ŃĆéµØéĶ┤©µŖźÕæŖµ¢╣µ│ĢÕÅ»ÕÅéńģ¦ICH Q3AÕÆīQ3B�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕ║öÕćŁĶ»üÕēéÕ×ŗŃĆüõ║¦Õōüńē╣ńé╣ńŁēĶ«ŠńĮ«ńøĖÕ«£ńÜäĶ┤©µÄ¦ķĪ╣ńø«ÕÆīÕē¢µ×ÉĶ”üķóå�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕģ│õ║Äõ╗źń¦»ń┤»µĢ░µŹ«õĖ║ńø«ńÜä�’╝ī�’╝īõĮåõĖŹõĮ£õĖ║ÕłČÕēéµöŠĶĪīµØĪõ╗ČńÜ䵯ƵĄŗķĪ╣ńø«�’╝ī�’╝īÕ║öõ║łõ╗źµ│©µśÄ�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕ£©ĶŹ»ÕōüÕ╝ĆÕÅæÕłØµ£¤�’╝ī�’╝īõĖŹķ£ĆĶ”üµÅÉõ║żÕæ©Õģ©Õ«īµĢ┤ńÜäÕē¢µ×ÉĶ”üķóåķ¬īĶ»üĶĄäµ¢Ö�’╝ī�’╝īõĮåĶć│Õ░æÕ║öµÅÉõŠøĶ”üķóåńÜäõĖōÕ▒׵ƦŃĆüĶ┐ģķƤÕ║”ńŁēĶ”üÕ«│ķĪ╣ńø«ńÜäķ¬īĶ»üõ┐Īµü»�ŃĆéŃĆéŃĆéŃĆé�ŃĆéµÅÉõŠøĶ”üÕ«│ńĀöń®Čµē╣µ¼Ī(Õ”éńö©õ║ĵĖģķØֵƦńĀöń®ČŃĆüń©│Õø║µĆ¦ńĀöń®ČŃĆüõĖ┤Õ║ŖńĀöń®ČńŁē)ńÜäńŻ©ń╗āµŖźÕæŖõ╣”�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕ║öµÅÉõŠøÕłČÕēéķÖŹĶ¦ŻķĆöÕŠäŃĆüķÖŹĶ¦Żõ║¦ÕōüńÜäĶĄĘµ║ÉńĀöń®ČµĢłµ×£�ŃĆéŃĆéŃĆéŃĆé��’╝¤�’╝¤�’╝¤�’╝¤�’╝¤Õł╣µÅĪĶ¬īCH Q3B�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
ŌśåŌśåń©│Õø║µĆ¦
Õ║öµÅÉõŠøÕłČÕēéń©│Õø║µĆ¦ńĀöń®ČĶĄäµ¢Ö�’╝ī�’╝īÕłŚµśÄµÄźń║│ńÜäÕē¢µ×ÉĶ”üķóå�’╝ī�’╝īÕÅ»ńö©ÕłŚĶĪ©ÕĮóÕ╝ŵÅÉõ║żµÄźĶĪ©µĆ¦µĀĘÕōü(Õ”éÕŖ©ńē®ĶŹ»ńÉåµ»ÆńÉåÕŁ”ńĀöń®ČµĀĘÕōüŃĆüµŗ¤ńö©õ║ÄõĖ┤Õ║ŖĶ»Ģķ¬īńÜäµĀĘÕōü)ńÜäĶĄĘµ║ɵĢ░µŹ«ÕÅŖÕģČõ╗¢µö»µīüµĆ¦ń©│Õø║µĆ¦ńĀöń®ČĶĄäµ¢Ö�’╝ī�’╝īÕ║öµÅÉõŠøĶ”üÕ«│ķĪ╣ńø«ńÜäõ╗ŻĶĪ©µĆ¦ÕøŠĶ░▒�ŃĆéŃĆéŃĆéŃĆé�ŃĆéń©│Õø║µĆ¦µĢ░µŹ«Õ║öĶāĮµö»µīüÕłČÕēéńÜäńÉåÕī¢ÕÅéµĢ░Õ£©Õ”äµā│ńÜäõĖ┤Õ║ŖńĀöń®ČµŚČõ╗ŻÕłćÕÉłĶ”üµ▒é�’╝ī�’╝īĶŗźµś»Õ”äµā│ńÜäĶ»Ģķ¬īÕ橵£¤µ×üń¤Ł�’╝ī�’╝īÕÅ»µÅÉõŠøµ£ēķÖÉńÜäµö»µīüµĆ¦ń©│Õø║µĆ¦µĢ░µŹ«�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
ŌśåŌśåÕīģĶŻģÕÆīĶ┤«ÕŁśµØĪõ╗Č
Õ║öÕłŚµśÄńø┤µÄźµÄźĶ¦”ÕīģĶŻģĶ┤©µ¢ÖńÜäõ┐Īµü»ÕÆīĶ┤«ÕŁśµØĪõ╗Č�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕģ│õ║ĵ¢░Ķ┤©µ¢ÖŃĆüµ¢░ń╗ōµ×äŃĆüµ¢░ńö©ķĆöńÜäĶŹ»ÕīģµØÉ�’╝ī�’╝īķ£ĆµÅÉõŠøõ┐Īµü»Õ╣ČÕćŁĶ»üĶ”üµ▒éõĖŠĶĪīÕģ│Ķüöńö│µŖź�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
ŌśåŌśåÕģČõ╗¢
Õģ│õ║ÄõĖ┤Õ║Ŗķ£ĆĶ”üõĖŠĶĪīķģŹõ╝ŹõĮ┐ńö©ÕÅŖµ£ēńē╣µ«ŖõĮ┐ńö©Ķ”üµ▒éńÜäÕłČÕēéÕ║öµÅÉõŠøńøĖÕģ│ń©│Õø║µĆ¦Õ«×ķ¬īµĢłµ×£�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
Ōśåµģ░ĶŚēÕēéõ┐Īµü»
Õ”éõĖ┤Õ║ŖĶ»Ģķ¬īĶ«ĪÕłÆõĖŁķ£ĆõĮ┐ńö©µģ░ĶŚēÕēé�’╝ī�’╝īÕ║öµÅÉõŠøµģ░ĶŚēÕēéńÜäÕżäµ¢╣ŃĆüńö¤õ║¦ÕĘźĶē║ÕÅŖńö¤õ║¦ÕÄéńÜäńøĖÕģ│õ┐Īµü»ŃĆüĶ┤©ķćÅµÄ¦ÕłČÕÆīńŻ©ń╗āµĢłµ×£ńŁēńĀöń®ČĶĄäµ¢Ö�ŃĆéŃĆéŃĆéŃĆé�ŃĆé
ķÖä’╝Ü
Õī¢ÕŁ”ĶŹ»ÕōüŌģĀµ£¤õĖ┤Õ║ŖĶ»Ģķ¬īńö│Ķ»ĘĶŹ»ÕŁ”ńĀöń®Čõ┐Īµü»µ▒ćµĆ╗ĶĪ©
1.Õ¤║µ£¼õ┐Īµü»

2.Ķ┤©µ¢ÖĶŹ»õ┐Īµü»
3.ÕłČÕēéõ┐Īµü»
ń¼öĶĆģµä¤õ╝ż’╝Ü
Õ£©ÕüÜń¦æńĀöńÜäÕÄåń©ŗõĖŁ�’╝ī�’╝īµ£ēµŚČµłæõ╗¼õ╝ÜõĖŹµĖģµÖ░õĖĆõ╗Čõ║ŗµāģń®Čń½¤ÕüÜÕł░õ╗Ćõ╣łµ░┤Õ╣│�’╝ī�’╝īµēŹµś»ń£¤µŁŻńÜäķĆéÕ║”...ķØóõĖ┤õ╗ŖÕż®ńÜäÕ«ĪĶ»äµĆüÕŖ┐�’╝ī�’╝īõĖ║õ║åÕ░ĮÕÅ»ĶāĮńÜäń¤źĶČ│Õ«ĪĶ»äõĖŁÕ┐āńÜäĶ”üµ▒é�’╝ī�’╝īµłæõ╗¼ÕŠĆÕŠĆõ╝ÜÕ»╣µ¤ÉõĖĆķśČµ«ĄńÜäĶŹ»ÕŁ”ńĀöń®Č�’╝ī�’╝īÕüÜÕć║Õż¬Ķ┐ćńÜäµŖĢÕģź�ŃĆéŃĆéŃĆéŃĆé�ŃĆéÕ»╣µŁż�’╝ī�’╝īµŚĀÕÅ»ÕÄÜķØ×�’╝ī�’╝īõ║ŗÕ«×ķØóõĖ┤ÕēŹµ£¤ÕĘ©ķóØńÜäµŖĢÕģź�’╝ī�’╝īõĖŹÕÅ»ńö▒õ║ÄĶ┐ÖŌĆ£õĖĆńé╣ńé╣ŌĆØńÜäĶŹ»ÕŁ”ńĀöń®ČµĢ░µŹ«�’╝ī�’╝īĶĆīÕ╗Čõ╝ĖµĢ┤õĖ¬ķĪ╣ńø«ń│╗ń╗¤ńÜäÕÄåń©ŗ...õĮåń¼öĶĆģõ╗źõĖ║�’╝ī�’╝īÕż¬Ķ┐ćńÜäńĀöÕÅæµŖĢÕģźõĖŹõĮåµś»Õ»╣õ║║ÕŖøŃĆüńē®ÕŖøńŁēĶĄäµ║ÉńÜäķō║Õ╝Ā�’╝ī�’╝īĶāīÕÉÄķĆÅķ£▓ńÜäµø┤µś»Õ»╣ķĪ╣ńø«ŃĆüÕ»╣ń¦æńĀöŃĆüÕ»╣ĶŹ»ńē®ńĀöÕÅæńÜ䵜ÄńĪ«õĖŹµĢĘ�ŃĆéŃĆéŃĆéŃĆé�ŃĆéķÆłÕ»╣µ¤ÉõĖĆńĀöÕÅæķśČµ«Ą�’╝ī�’╝īµłæķ£ĆĶ”üĶÄĘÕŠŚµĆĵĀĘńÜäµĢ░µŹ«µö»µīü�’╝ī�’╝īµłæÕÅłĶ”üµŖĢÕģźÕżÜÕż¦Õ«ŚńÜäĶ»Ģķ¬īµĢ░ńø«�’╝ī�’╝īĶ»Ėõ║æõ║æń▒╗ńÜäķŚ«ķóś�’╝ī�’╝īĶć│Õ┐āķ£ĆĶ”üÕżÜÕżÜÕ»╣Ķć¬ÕĘ▒µÅÉķŚ«�’╝ī�’╝īńäČÕÉÄÕÄ╗Ķ¦ŻÕå│Õ«ā�’╝ī�’╝īÕĮōń¢æķŚ«ĶČŖµØźĶČŖÕ░æõ╣ŗµŚČ�’╝ī�’╝īńøĖõ┐ĪµēŹõ╝ÜÕ»╣ķĪ╣ńø«ńÜ䵜ÄńĪ«µø┤õĖ║ķĆÅÕĮ╗�’╝ī�’╝īõ╣¤Ķ«Ėķ鯵ŚČ�’╝ī�’╝īµēŹÕŹÄń£¤µŁŻń”╗ń¦æÕŁ”µø┤Ķ┐øõĖƵŁźÕɦ’╝üµŗÖĶ¦ü~
ÕÅéĶĆā’╝Ü
1.https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm071597.pdf
2.https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm070567.pdf
3.https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm078933.pdf
4.https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm070273.pdf
5.https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm229175.pdf
6. Õī¢ÕŁ”ĶŹ»ńē®(Ķ┤©µ¢ÖĶŹ»ÕÆīÕłČÕēé)ń©│Õø║µĆ¦ńĀöń®ČµēŗĶē║µīćÕ»╝ÕÄ¤ÕłÖ(20150205)
7. Õī¢ÕŁ”ĶŹ»ńē®Ķ┤©µ¢ÖĶŹ»ÕłČÕżćÕÆīń╗ōµ×äńĪ«Ķ»üńĀöń®ČµēŗĶē║µīćÕ»╝ÕÄ¤ÕłÖ(20070823)
8. Õī¢ĶŹ»ĶŹ»ÕōüńĀöń®ČĶĄäµ¢ÖÕÅŖÕøŠĶ░▒ń£¤Õ«×µĆ¦ķŚ«ķóśÕłżµ¢ŁµĀćÕćå(20100510)
9. ĶŹ»ńē®ŌģĀµ£¤õĖ┤Õ║ŖĶ»Ģķ¬īµ▓╗ńÉåµīćÕ»╝ÕÄ¤ÕłÖ(Ķ»ĢĶĪī)(20111207)
10. CNKI-ń½ŗÕ╝éĶŹ»ĶŹ»ÕŁ”ńĀöń®ČńÜäńē╣ńé╣ÕÅŖµēŗĶē║µĆØķćÅ�ŃĆéŃĆéŃĆéŃĆé�ŃĆé

Õłåõ║½Õł░’╝Ü



















