еҲ¶иҚҜиЎҢдёҡе…ідәҺBTDеҰ„жғізҡ„дёүеӨ§з–‘й—®
2012е№ҙ7жңҲпјҢ�пјҢеңЁгҖҠзҫҺеӣҪйЈҹзү©иҚҜе“ҒжІ»зҗҶеұҖжё…йқҷдёҺз«ӢејӮжі•жЎҲгҖӢпјҲFDASIAпјүиҺ·еҫ—еӣҪдјҡжү№еҮҶд№ӢеҗҺпјҢ�пјҢзӘҒз ҙжҖ§з–—жі•и®Өе®ҡпјҲBTDпјүжңҖе…Ҳз”ҹж•ҲгҖӮгҖӮ�гҖӮ�гҖӮ新规дёәз”ЁжқҘжІ»з–—дёҘйҮҚз–ҫз—…жҲ–иҖ…еҚұеҸҠз”ҹе‘Ҫзҡ„з–ҫз—…зҡ„иҚҜзү©жҸҗдҫӣдәҶж–°зҡ„иө„ж ји®Өе®ҡпјҢ�пјҢеҲ¶иҚҜе…¬еҸёиҰҒиҺ·еҫ—иҝҷз§Қиө„ж јпјҢ�пјҢйңҖиҰҒйҖҡиҝҮиө·жәҗдёҙеәҠиҜҒжҚ®жү№жіЁпјҢ�пјҢе…¶з ”еҸ‘зҡ„ж–°иҚҜжҳҫзӨәеҮәдәҶжҜ”зҺ°жңүиҚҜзү©жңүжҳҫзқҖж”№е–„зҡ„жІ»з–—ж•ҲжһңгҖӮгҖӮ�гҖӮ�гҖӮ
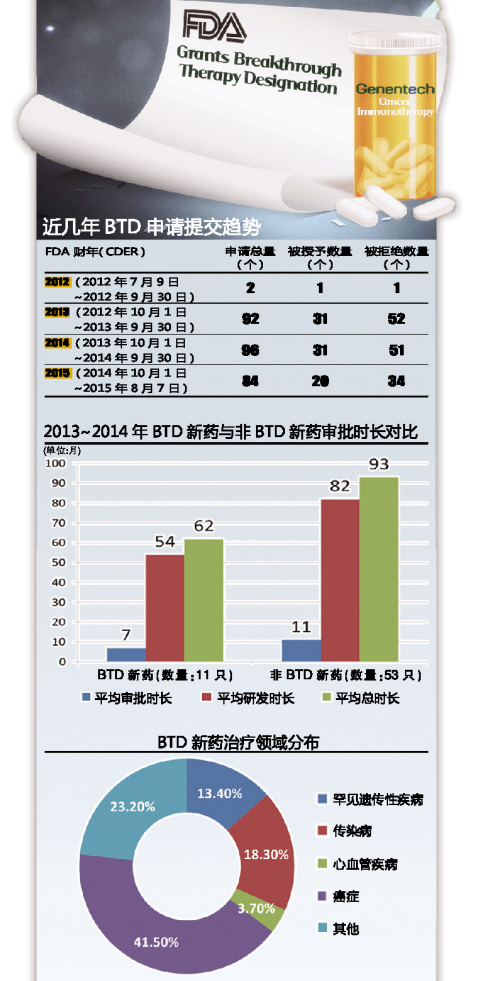
еңЁFDAеҮәеҸ°зҡ„дёҖзі»еҲ—еҠ йҖҹиҚҜзү©ејҖеҸ‘е’Ңжү№еҮҶеҰ„жғідёӯпјҢ�пјҢBTDжңҖеҲқзңӢдјјжҳҜдёҖдёӘзӣёеҪ“дёҚиө·зңјзҡ„еўһиЎҘпјҢ�пјҢеӨ–з•ҢеҜ№е…¶жүҖзҲҶеҸ‘еҪұе“Қзҡ„йў„жңҹ并дёҚеӨ§пјҢ�пјҢеңЁжңүдәӣдәәзңӢжқҘпјҢ�пјҢеҲ¶иҚҜиЎҢдёҡжҜҸе№ҙжҸҗеҮәзҡ„зӘҒз ҙжҖ§з–—жі•и®Өе®ҡз”іиҜ·д№ҹи®ёе°‘еҲ°еҸӘжңүдёӨдёү件гҖӮгҖӮ�гҖӮ�гҖӮ然иҖҢпјҢ�пјҢеҮӯиҜҒFDAе®Јеёғзҡ„2013е№ҙиЎҢдёҡжҢҮеҜјжҠҘе‘ҠпјҢ�пјҢеңЁиҝҷйЎ№з«Ӣжі•з”ҹж•Ҳд№ӢеҗҺзҡ„зҹӯзҹӯдёҖе№ҙж—¶й—ҙйҮҢпјҢ�пјҢиҝҷдёҖж•°еӯ—дёҠж¶ЁдәҶеҚҒеҖҚгҖӮгҖӮ�гҖӮ�гҖӮзҺ°еңЁпјҢ�пјҢеңЁBTDеҰ„жғіиҝӣе…Ҙзҡ„第дёүдёӘе№ҙеӨҙпјҢ�пјҢзҺ°е®һж•°еӯ—жҜ”жңҖеҲқзҡ„йў„и®Ўй«ҳдәҶ30~40еҖҚгҖӮгҖӮ�гҖӮ�гҖӮ
Q1:BTDе®Ўжү№жҸҗйҖҹжҳҜеҗҰе®һиҮіеҗҚеҪ’�пјҹпјҹ�пјҹпјҹ�пјҹ
д»Һв… жңҹдёҙеәҠиҜ•йӘҢйҳ¶ж®өжңҖе…ҲпјҢ�пјҢBTDе°ұдёәй«ҳж•Ҳзҡ„иҚҜзү©ејҖеҸ‘жҸҗдҫӣдәҶжңүеҠӣеҗҺжҸҙпјҢ�пјҢFDAеҗ„йғЁеҲҶеңЁи·ЁеӯҰ科зҡ„еҚҸеҗҢеҠ е…Ҙдёӯеҗ„еҸёе…¶иҒҢ
еҲқзңӢиө·жқҘпјҢ�пјҢBTDдјјд№Һе°ұжҳҜFDAеҝ«йҖҹе®Ўжү№йҖҡйҒ“пјҲFast Trackпјүзҡ„еҸҰеӨ–дёҖдёӘзҝ»зүҲгҖӮгҖӮ�гҖӮ��пјҹпјҹ�пјҹпјҹ�пјҹз„–еҒ•з¬ҲЗ–жң—иҚҲDAе»әи®ҫзҡ„дёҖдёӘиҮӘеҠӣзҡ„иҚҜе“Ғе®ЎиҜ„иө„ж јеҰ„жғігҖӮгҖӮ�гҖӮ�гҖӮдёҚиҝҮпјҢ�пјҢFDAеҫҲеҝ«еҸ‘жҳҺпјҢ�пјҢBTDжүҖеёҰжқҘзҡ„еҲ©зӣҠиҰҒжҜ”еҝ«йҖҹе®Ўжү№йҖҡйҒ“и¶ҠеҸ‘жҷ®йҒҚгҖӮгҖӮ�гҖӮ�гҖӮд»Һв… жңҹдёҙеәҠиҜ•йӘҢйҳ¶ж®өжңҖе…ҲпјҢ�пјҢBTDе°ұдёәй«ҳж•Ҳзҡ„иҚҜзү©ејҖеҸ‘жҸҗдҫӣдәҶејәжңүеҠӣзҡ„жҢҮеҜјпјҢ�пјҢе°Өе…¶жҳҜе…¶иҜҶеҲ«еҮәдәҶдёҙеәҠиҜ•йӘҢи®ҫи®ЎеҸ–еҫ—зҡ„еүҚиҝӣпјҲеҘҪжҜ”йЎәеә”жҖ§дёҙеәҠиҜ•йӘҢпјүпјҢ�пјҢд»ҘеҸҠдёҖзӣҙз”ҹй•ҝд№Ӣдёӯзҡ„жүӢиүәпјҲеҘҪжҜ”йҷӘеҗҢејҸиҜҠж–ӯпјүгҖӮгҖӮ�гҖӮ�гҖӮ
жңҖеҖјеҫ—жіЁйҮҚзҡ„жҳҜпјҢ�пјҢBTDеҗ‘дёҙеәҠиҜ•йӘҢжҸҗеҖЎиҖ…жҸҗдҫӣдәҶFDAзҡ„е…Ғи®ёпјҢ�пјҢиҝҷдёҖзӮ№иў«и§ҶдёәFDAеңЁи·ЁеӯҰ科зҡ„еҚҸеҗҢеҠ е…Ҙдёӯе°ҶдјҡеҒҡеҲ°еҗ„еҸёе…¶иҒҢпјҢ�пјҢиҝҷз§ҚеҠ е…ҘдёҚдҪҶд»…жҳҜеңЁFDAйғЁеҲҶеұӮйқўпјҢ�пјҢ并且иҝҳи·Ёи¶ҠдәҶеҗ„зә§жІ»зҗҶеұӮгҖӮгҖӮ�гҖӮ�гҖӮ
然иҖҢпјҢ�пјҢFDAе’ҢдёҙеәҠиҜ•йӘҢжҸҗеҖЎиҖ…еҫҲеҝ«е°ұйҒӯйҒҮеҲ°дәҶжҢ‘жҲҳгҖӮгҖӮ�гҖӮ�гҖӮе°ұFDAж–№йқўжқҘиҜҙпјҢ�пјҢйқўдёҙз”іиҜ·еӨӘеӨҡгҖҒж—¶й—ҙеӨӘзҹӯзҡ„й—®йўҳгҖӮгҖӮ�гҖӮ�гҖӮзҺ°еңЁпјҢ�пјҢFDAи§ЈеҶізҡ„жӯҘдјҗе°ұжҳҜе…Ғи®ёз»ҷдәҲдёҖе®ҡзҡ„ж— йӮӘжҖ§гҖӮгҖӮ�гҖӮ�гҖӮFDAе®ЎиҜ„иҒҢе‘ҳиөһжҲҗпјҢ�пјҢеҲ¶иҚҜе…¬еҸёеңЁжҸҗдәӨз”іиҜ·ж—¶пјҢ�пјҢеҸҜд»ҘжҸҗдҫӣиҫғе°‘зҡ„зЁіеӣәжҖ§ж•°жҚ®пјҢ�пјҢ他们еңЁе®ЎиҜ„е‘ЁжңҹеҶ…д№ҹеҸҜд»ҘжҺҘеҸ—з ”з©¶ж•°жҚ®зҡ„дҝ®ж”№пјҢ�пјҢ并еўһж·»дә§е“ҒдёҠеёӮй”Җе”®д№ӢеҗҺзҡ„зӣёе…іе…Ғи®ёгҖӮгҖӮ�гҖӮ�гҖӮFDAдҪ“зҺ°пјҢ�пјҢе…¶ж„ҝж„ҸеңЁиҝҷдёӘе®ЎиҜ„зЁӢеәҸдёӯиҝӣдёҖжӯҘжӢ“еұ•иҝҷз§ҚдҫҝеҪ“жҖ§гҖӮгҖӮ�гҖӮ�гҖӮ
еңЁйҒӯеҲ°жӢ’з»қзҡ„BTDз”іиҜ·дёӯпјҢ�пјҢзәҰиҺ«72%дёҺиҜ•йӘҢи®ҫи®ЎжҲ–еү–жһҗй—®йўҳжңүе…ігҖӮгҖӮ�гҖӮ�гҖӮиҝҷжү№жіЁпјҢ�пјҢиҜ•йӘҢжҸҗеҖЎиҖ…еңЁжҸҗдәӨз”іиҜ·д№ӢеүҚпјҢ�пјҢеҝ…йңҖеңЁдёҺFDAзҡ„зӣёеҗҢдәӨжөҒдёҠдёӢеҠҹеӨ«гҖӮгҖӮ�гҖӮ�гҖӮдёҺFDAзӣёе…іиҜ„е®ЎйғЁеҲҶдёҫиЎҢйқһжӯЈејҸзҡ„жҺўи®ЁпјҢ�пјҢеҸҜиғҪдјҡжңүеҠ©дәҺиҜ•йӘҢжҸҗеҖЎиҖ…жӣҙеҘҪгҖҒжӣҙж—©ең°зӣёиҜҶFDA究з«ҹйңҖиҰҒд»Җд№Ҳж ·зҡ„ж•°жҚ®гҖӮгҖӮ�гҖӮ�гҖӮ
еңЁзҫҺеӣҪжҷәеә“вҖңеёғйІҒйҮ‘ж–ҜеӯҰдјҡвҖқжңҖиҝ‘дёҫиЎҢзҡ„иҒҡдјҡдёҠпјҢ�пјҢжңүдәәжҢҮеҮәпјҢ�пјҢиҝҷз§Қж—©жңҹзҡ„зӣёеҗҢдәӨжөҒж—ўеҸҜд»ҘеўһејәBTDз”іиҜ·зҡ„еҸҜиЎҢжҖ§пјҢ�пјҢеҸҲеҸҜд»Ҙй•Ңжұ°йӮЈдәӣжҜ«ж— ж„Ҹд№үзҡ„з”іиҜ·ж•°зӣ®гҖӮгҖӮ�гҖӮ�гҖӮ
е…ідәҺдёҙеәҠиҜ•йӘҢжҸҗеҖЎиҖ…иҖҢиЁҖпјҢ�пјҢжҢ‘жҲҳеҲҷеңЁдәҺе…¶иҮӘиә«иө„жәҗж–№йқўгҖӮгҖӮ�гҖӮ�гҖӮеҮӯиҜҒBTD规еҲҷпјҢ�пјҢдә§е“Ғзҡ„дёҠеёӮй”Җе”®з”іиҜ·еңЁв…ЎжңҹиҜ•йӘҢйҳ¶ж®өд№ӢеҗҺиў«жҸҗдәӨпјҢ�пјҢиҝҷжҜ”еҸӨжқҝзҡ„ж–°иҚҜдёҠеёӮе…Ғи®ёз”іиҜ·пјҲNDAпјүжҸҗж—©дёӨе№ҙпјҢ�пјҢиҝҷз§Қжғ…еҪўдјҡз”ұдәҺеҲ¶иҚҜе…¬еҸёеңЁе…ЁзҗғеӨҡдёӘеёӮеңәеҗҢж—¶з”іиҜ·иҖҢиҝӣдёҖжӯҘйҮҚеӨ§еҢ–пјҲеҢ…жӢ¬еӨ–жҙӢзҫҒзі»жңәжһ„еҜ№з”ҹдә§зҺ°еңәдёҫиЎҢжЈҖжҹҘпјүгҖӮгҖӮ�гҖӮ�гҖӮеҜ№з”ҹзү©еҲ¶еүӮзҡ„з”іиҜ·жқҘиҜҙпјҢ�пјҢBTDеҸҜиғҪиҝҳж¶үеҸҠеҲ°еҲ¶иҚҜе…¬еҸёиҰҒд»Ҙи¶ҠеҸ‘жҷ®йҒҚзҡ„ж ҮеҮҶ规иҢғжҸҗдәӨз”іиҜ·пјҢ�пјҢиҝҷдәӣж ҮеҮҶ规иҢғеңЁйЎ№зӣ®еҗҺжңҹйҳ¶ж®өпјҲд»ҺдёҙеәҠиҜ•йӘҢеҹәең°зҡ„жҺЁеҮәпјҢ�пјҢд»ҘеҸҠиҪ¬з§»еҲ°е•Ҷдёҡз”ҹдә§еҹәең°зҡ„дёҠеёӮй”Җе”®д№ӢеҗҺпјүдјҡеҸҳеҫ—и¶ҠеҸ‘дёҘй…·гҖӮгҖӮ�гҖӮ�гҖӮеӣ жӯӨпјҢ�пјҢеҲ¶иҚҜе…¬еҸёеҝ…йңҖеңЁз”іиҜ·дёӯеҢ…жӢ¬дә§е“ҒиҺ·жү№д№ӢеҗҺзҡ„з”ҹе‘Ҫе‘ЁжңҹжІ»зҗҶеҰ„жғігҖӮгҖӮ�гҖӮ�гҖӮ
иҝҷеҸҜиғҪеј•еҸ‘зҡ„ж•Ҳжһңе°ұжҳҜпјҢ�пјҢиӢҘжҳҜеҲ¶иҚҜе…¬еҸёиҝҮдәҺйў‘д»Қең°еҜ№жҪңеңЁзҡ„зӘҒз ҙжҖ§иҚҜзү©жҠ•дёӢиөҢжіЁпјҢ�пјҢиҝҷе°ұдјҡи®©иҮӘиә«зҡ„иө„жәҗеӨ„дәҺеӨӘиҝҮдё»иҰҒзҠ¶жҖҒгҖӮгҖӮ�гҖӮ�гҖӮFDAд№ҹжӢ…еҝғиҝҷдёҖзӮ№пјҢ�пјҢз”ұдәҺBTDеҰ„жғіе№ҝеҸ—жҺҘеҫ…пјҢ�пјҢдҪҶз«Ӣ法并没жңүдёәзҹҘи¶іжӣҙеӨҡзҡ„з”іиҜ·йңҖжұӮжҸҗдҫӣи¶іеӨҹзҡ„иө„йҮ‘ж”ҜжҢҒгҖӮгҖӮ�гҖӮ�гҖӮ
Q2:BTDжҳҜеҗҰеҖјеҫ—иҚҜдјҒе®һйӘҢ�пјҹпјҹ�пјҹпјҹ�пјҹ
2013~2014е№ҙпјҢ�пјҢ BTDж–°иҚҜзҡ„е№іеқҮжҖ»ејҖеҸ‘ж—¶й—ҙжҜ”йқһBTDж–°иҚҜе°‘2.5е№ҙ
иӢҘжҳҜBTDеёҰеҫҖиҝ”жҠҘпјҢ�пјҢдёҙеәҠиҜ•йӘҢжҸҗеҖЎиҖ…дјҡиҺ·еҫ—д»Җд№Ҳ�пјҹпјҹ�пјҹпјҹ�пјҹе№іеқҮжқҘиҜҙпјҢ�пјҢ2013~2014е№ҙпјҢ�пјҢж–°еҲҶеӯҗе®һдҪ“пјҲNMEпјүиҚҜзү©жү№еҮҶзҡ„BTDдә§е“ҒпјҲж•°зӣ®дёә11еҸӘпјүзҡ„жҖ»ејҖеҸ‘ж—¶й—ҙиҰҒжҜ”йқһBTDж–°еҲҶеӯҗе®һдҪ“пјҲж•°зӣ®дёә53еҸӘпјүе°‘2.5е№ҙгҖӮгҖӮ�гҖӮ��пјӣ�пјӣ�пјӣ�пјӣ�пјӣз—Әжө е…«жӢ…�пјҢBTDиҮӘе·ұеёҰжқҘдәҶиҝ‘3.5е№ҙзҡ„е®Ўжү№ж—¶й—ҙдјҳеҠҝгҖӮгҖӮ�гҖӮ�гҖӮ
е°ұBTDж–№йқўжқҘиҜҙпјҢ�пјҢиҝҷз§ҚеҲқжңҹзҡ„дҪ“зҺ°е№¶жІЎжңүиў«жҠ•иө„з•ҢжүҖеҝҪи§ҶгҖӮгҖӮ�гҖӮ�гҖӮ2014е№ҙпјҢ�пјҢжҠ•е…Ҙз”ҹе‘Ҫ科еӯҰе…¬еҸёзҡ„иө„йҮ‘еҚ еҚұе®іиө„жәҗжҠ•иө„жҖ»йўқзҡ„18%пјҢ�пјҢе…үжҳҜеҚұе®іиө„жәҗ家е°ұеҜ№з§ҒиҗҘз”ҹзү©жүӢиүәе…¬еҸёжҠ•е…ҘдәҶ60дәҝзҫҺе…ғгҖӮгҖӮ�гҖӮ�гҖӮзҫҺеӣҪеӣҪдјҡдёҫиЎҢзӣёе…іиҜқйўҳи®Ёи®әж—¶з§°пјҢ�пјҢиҝҷз§Қйқўеҗ‘з”ҹзү©жүӢиүәе’ҢеҲ¶иҚҜйўҶеҹҹзҡ„з§ҒиҗҘжҠ•иө„и¶ӢеҠҝжҳҜиө·еҠІзҡ„зҫҒзі»е’ҢеҶіи®®жғ…еҪўжүҖеёҰжқҘзҡ„ж•ҲжһңгҖӮгҖӮ�гҖӮ�гҖӮ
дёҺжӯӨеҗҢж—¶пјҢ�пјҢе®ҪеӨ§жӮЈиҖ…иӮҜе®ҡдјҡеҜ№BTDеҰ„жғіеҸ–еҫ—зҡ„ж•Ҳжһңж„ҹеә”е…ҙеҘӢпјҡе·ІеҫҖдёүе№ҙжқҘпјҢ�пјҢеҢ»иҚҜиЎҢдёҡжҸҗдәӨдәҶ300еӨҡ件иө„ж ји®Өе®ҡз”іиҜ·пјҢ�пјҢжңүеҮҢй©ҫ25еҸӘдә§е“ҒиҺ·еҫ—жү№еҮҶпјҢ�пјҢе…¶дёӯеҢ…жӢ¬еңЁFDA иҚҜзү©иҜ„д»·дёҺз ”з©¶дёӯеҝғпјҲCDERпјүе’Ңз”ҹзү©еҲ¶е“ҒиҜ„д»·дёҺз ”з©¶дёӯеҝғпјҲCBERпјүиҺ·еҫ—жү№еҮҶзҡ„NMEе’ҢеўһиЎҘеүӮпјҢ�пјҢиҝҷдәӣдә§е“ҒжЁӘи·ЁеӨҡдёӘжІ»з–—йўҶеҹҹгҖӮгҖӮ�гҖӮ�гҖӮ
Q3:BTDжҳҜеҗҰдёәе…¬дј—еә·еҒҘеёҰжқҘжӣҙеӨ§еҲ©зӣҠ�пјҹпјҹ�пјҹпјҹ�пјҹ
еңЁжӯӨжЎҶжһ¶дёӢпјҢ�пјҢFDAиғҪеӨҹж”ҜжҢҒд»Һж—©жңҹдёҙеәҠиҜ•йӘҢйҳ¶ж®өеҲ°дёҠеёӮй”Җе”®еҗҺеҚұе®іжІ»зҗҶйҳ¶ж®өзҡ„еҢ»иҚҜз«ӢејӮпјҢ�пјҢдҪҶйңҖжіЁйҮҚиө„жәҗжІ»зҗҶ
FDAжҢҮеҮәпјҢ�пјҢBTDзҡ„жҺЁеҠЁеҠӣдё»иҰҒжәҗдәҺиҚҜзү©з ”еҸ‘йўҶеҹҹжіӣиө·зҡ„еҮ дёӘж–°и¶ӢеҠҝпјҢ�пјҢеҘҪжҜ”еҲҶеӯҗйқ¶еҗ‘жІ»з–—иҚҜзү©зҡ„е…ҙиө·пјҢ�пјҢе…¶еҫҖеҫҖдёҺйҷӘеҗҢејҸиҜҠж–ӯе·Ҙе…·иҝһзі»еңЁдёҖиө·гҖӮгҖӮ�гҖӮ�гҖӮеңЁиҝҷдәӣйқ¶еҗ‘жІ»з–—иҚҜзү©дёӯпјҢ�пјҢжңүдёҖдәӣжҜ”зҺ°жңүиҚҜзү©жІ»з–—ж•ҲжһңжӣҙеӨ§пјҢ�пјҢиҝҷз§Қж•Ҳжһңз”ҡиҮіеңЁеҜ№дәәдҪ“жүҖеҒҡзҡ„иө·жәҗиҜ•йӘҢдёӯе°ұдҪ“зҺ°еҫ—еҫҲжҳҫзқҖгҖӮгҖӮ�гҖӮ�гҖӮиӢҘжҳҜеңЁиҚҜе“ҒејҖеҸ‘зҡ„ж—©жңҹйҳ¶ж®өпјҢ�пјҢе°ұиғҪи§ҶеҜҹеҲ°иҚҜзү©еңЁжІ»з–—дёҘйҮҚз–ҫз—…ж—¶зҲҶеҸ‘дәҶеҫҲеӨ§зҡ„ж•ҲжһңпјҢ�пјҢйӮЈд№ҲпјҢ�пјҢе®һйӘҢжӣҙй•ҝж—¶й—ҙгҖҒз¬јзҪ©еҸӨжқҝзҡ„иҜ•йӘҢе…ЁзЁӢзҡ„дёҙеәҠејҖеҸ‘еҰ„жғізңӢдёҠеҺ»д№ҹе°ұжҳҫеҫ—еӨҡдҪҷдәҶгҖӮгҖӮ�гҖӮ�гҖӮ
дёҺеҸӨжқҝе®Ўжү№зЁӢеәҸжүҖйңҖзҡ„ж—¶й—ҙзӣёжҜ”пјҢ�пјҢFDAеңЁе°Ҫж—©еҗ‘жӮЈиҖ…жҸҗдҫӣиҝңжҷҜзңӢеҘҪзҡ„иҚҜзү©ж–№йқўд№ҹйқўдёҙзқҖжҢ‘жҲҳпјҢ�пјҢе°Өе…¶жҳҜеңЁжӮЈиҖ…жІЎжңүе…¶е®ғиҚҜзү©еҸҜдҫӣдҪҝз”Ёзҡ„жғ…еҪўдёӢгҖӮгҖӮ�гҖӮ�гҖӮBTDдјјд№ҺжҳҜдёҖжқЎи¶ҠеҸ‘еҲҮеҗҲFDAиҮӘиә«дҪҝе‘Ҫзҡ„и·Ҝеҫ„пјҡдёҖж–№йқўиҰҒ�пјӣ�пјӣ�пјӣ�пјӣ�пјӣгҒҷиҜҳзӣ®еҲ°гҖӮ�пјҢеҸҰеӨ–дёҖж–№йқўиҰҒеўһиҝӣеҢ»з–—дә§е“Ғзҡ„ејҖеҸ‘гҖӮгҖӮ�гҖӮ�гҖӮBTDиҰҒжұӮеҲ¶иҚҜе…¬еҸёжҳҺзЎ®пјҢ�пјҢе…¶жүҖејҖеҸ‘зҡ„иҚҜзү©еңЁе…·жңүдёҙеәҠж„Ҹд№үзҡ„з»ҲзӮ№дёҠжҜ”зҺ°еңЁеҸҜдҫӣдҪҝз”Ёзҡ„иҚҜзү©дҪ“зҺ°еҮәе®һиҙЁжҖ§зҡ„дјҳеҠҝпјҢ�пјҢжӯӨдёҫж—ўжҸҗдҫӣдәҶиғңиҝҮзҺ°жңүиҚҜзү©зҡ„дјҳеҠҝпјҢ�пјҢеҸҲжҸҗдҫӣдәҶеҗҲзҗҶеұ•жңӣдёҙеәҠжІ»з–—еҲ©зӣҠзҡ„иҜҒжҚ®гҖӮгҖӮ�гҖӮ�гҖӮ
еӣ жӯӨпјҢ�пјҢиҝҷе°Ҷи®©FDAжӢҘжңүдёҖз§ҚжүӢж®өпјҢ�пјҢз”ЁжқҘи§ЈеҶіе…¶д»ҘжҳҜдёәзҡ„зҫҒзі»жӮ–и®әпјҡдёҖж–№йқўпјҢ�пјҢFDAеҜ№иҚҜзү©зҡ„иҜёеӨҡдёҚзЎ®е®ҡжҖ§ж„ҹеә”зғҰжҮ‘пјҢ�пјҢеҸҰдёҖж–№йқўпјҢ�пјҢе…¶еҸҲж„ҝж„ҸжҺҘеҸ—дёҖеҸӘиҚҜзү©еңЁиҺ·жү№д№ӢеүҚдҝқеӯҳзҡ„жҹҗдәӣжңӘзҹҘж•°пјҢ�пјҢиҝҷдёӨиҖ…д№Ӣй—ҙдҝқеӯҳзқҖеӣәжңүдҪҶеҚҙеҸҜд»ҘеҚҸи°ғзҡ„зҹӣзӣҫгҖӮгҖӮ�гҖӮ�гҖӮд»ҘеүҚпјҢ�пјҢFDAжҳҜеңЁиҚҜзү©ејҖеҸ‘зҡ„иҰҒе®іиҠӮзӮ№дёҫиЎҢжҢүжңҹе№Ійў„пјҢ�пјҢеҸҜжҳҜзҺ°еңЁпјҢ�пјҢFDAзҡ„е№Ійў„ж–№жі•иҪ¬еҗ‘дәҶвҖңе…Ёз¬јзҪ©гҖҒжүҖжңүжқЎзҗҶгҖҒж—¶ж—¶еҲ»еҲ»вҖқзҡ„еўһиҝӣжЎҶжһ¶гҖӮгҖӮ�гҖӮ�гҖӮйҖҡиҝҮиҝҷз§ҚеҒҡжі•пјҢ�пјҢFDAиғҪеӨҹж”ҜжҢҒд»Һж—©жңҹдёҙеәҠиҜ•йӘҢйҳ¶ж®өеҲ°дёҠеёӮй”Җе”®еҗҺеҚұе®іжІ»зҗҶйҳ¶ж®өзҡ„еҢ»иҚҜз«ӢејӮгҖӮгҖӮ�гҖӮ�гҖӮ
дёҚиҝҮпјҢ�пјҢиҰҒи®©иҝҷз§Қжғ…еҪўйЎәйҒӮе®һзҺ°пјҢ�пјҢжңүе…іж–№йқўеҝ…йңҖеңЁиө„жәҗеұӮйқўжҺҘзәіе“Қеә”зҡ„жӯҘдјҗпјҡиҰҒд№ҲдёҙеәҠиҜ•йӘҢжҸҗеҖЎиҖ…еҝ…йңҖй•Ңжұ°з”іиҜ·ж•°зӣ®пјҢ�пјҢжҸҗдәӨжӣҙе°‘жҲ–иҖ…жӣҙй«ҳиҙЁйҮҸзҡ„з”іиҜ·пјҢ�пјҢиҰҒд№ҲFDAйҖҡиҝҮжү©еӨ§BTDеҰ„жғіе’Ң收еҸ–дёҖе®ҡзҡ„з”ЁеәҰпјҢ�пјҢжү©еӨ§е…¶е®Ўжү№и§„жЁЎгҖӮгҖӮ�гҖӮ�гҖӮ

еҲҶдә«еҲ°пјҡ



















